B About logical models
Several logical models of cancer are used in this thesis and some additional descriptive elements about them are given below.
B.1 Generic logical model of cancer pathways
For this thesis, a published Boolean model from (Fumia and Martins 2013) has first been used to illustrate our PROFILE methodology. This regulatory network summarizes several key players and pathways involved in cancer mechanisms such as RTKs, PI3K/AKT, WNT/\(\beta\)-catenin, TGF-\(\beta\)/Smads, Rb, HIF-1, p53 and ATM/ATR. An input node Acidosis has been added, along with an output node Proliferation used as a readout for the activity of any of the cyclins (CyclinA, CyclinB, CyclinD and CyclinE). This slightly extended model contains 98 nodes and 254 edges and its inputs are Acidosis, Nutrients, Growth Factors (GFs), Hypoxia, TNFalpha, ROS, PTEN, p14ARF, GLI, FOXO, APC and MAX. Its outputs are Proliferation, Apoptosis, DNA_repair, DNA_damage, VEGF, Lactic_acid, GSH, GLUT1 and COX412.
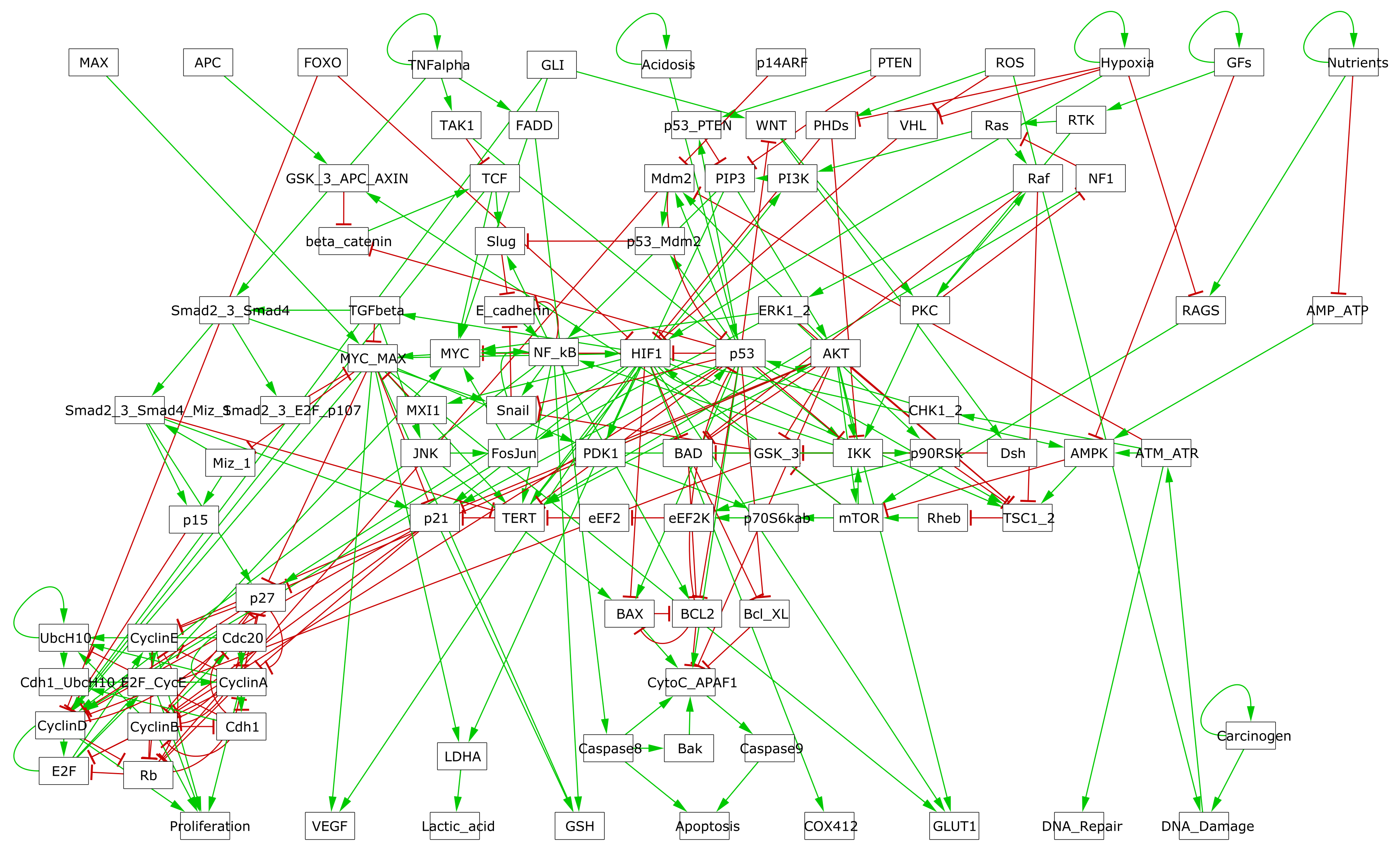
Figure B.1: GINsim representation of the logical model described in Fumia and Martins (2013).
B.2 Extended logical model of cancer pathways
Another logical model of similar size and scope was also used, primarily for the study of treatment responses. This model was built by Loïc Verlingue, a medical oncologist and member of the laboratory and preliminary versions of the model are described in Verlingue, Dugourd, et al. (2016) and Verlingue, Calzone, et al. (2016). One of the interests of this model is that it has been designed with a more clinical perspective, notably centred on the response to MTOR inhibitors. In addition, it presents more biological read-outs used for interpretation, and we will use mainly Proliferation (also called G1_S in the model files to designate the associated stage of the cell cycle), Apoptosis and Quiescence in particular. In addition, being able to discuss and collaborate directly with the model autor has helped to avoid potential errors in use.
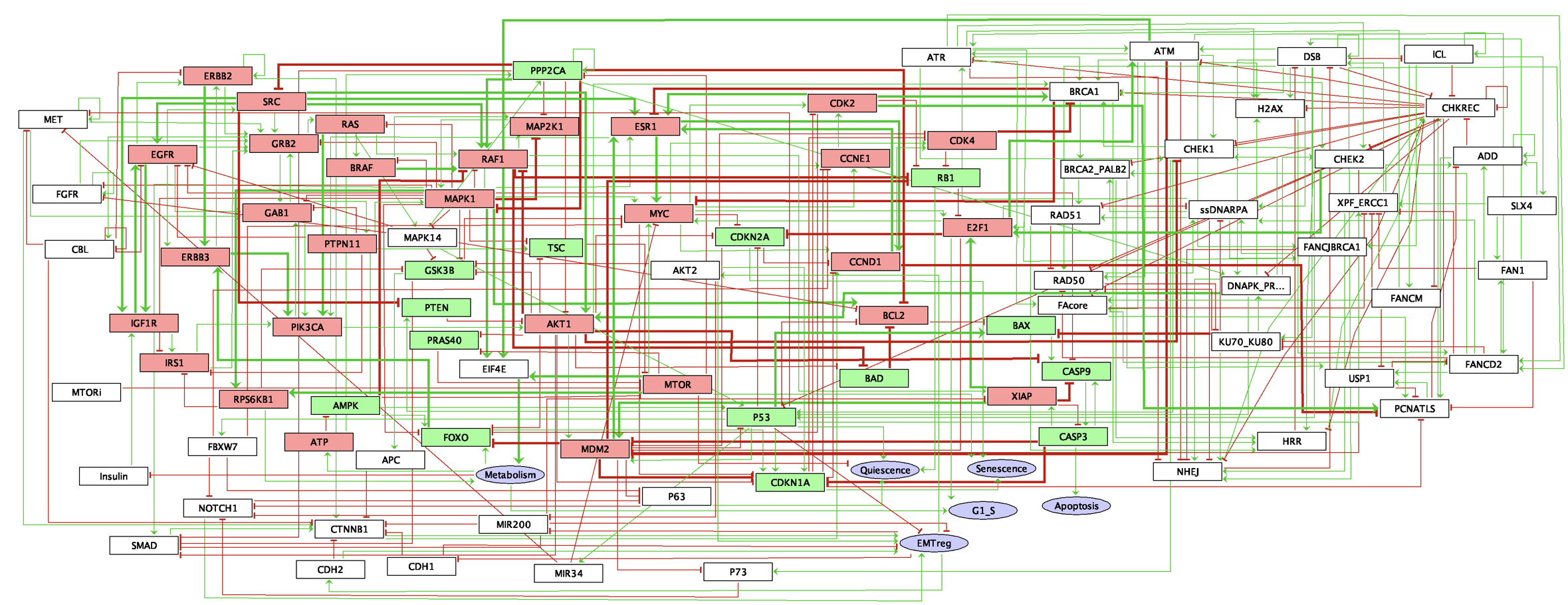
Figure B.2: GINsim representation of the 'Verlingue' logical model described in Verlingue, Calzone, et al. (2016).
B.3 Logical model of BRAF pathways in melanoma and colorectal cancer
Here are some details about the regulations represented in Figure 6.4. The MAPK pathway encompasses three families of protein kinases: RAF, MEK, ERK. If RAF is separated into two isoforms, CRAF and BRAF, the other two families MEK and ERK are represented by a single node. When BRAF is inhibited, ERK can still be activated through CRAF, and BRAF binds to and phosphorylates MEK1 and MEK2 more efficiently than CRAF (Wellbrock, Karasarides, and Marais 2004), especially in his V600E/K mutated form. When PI3K/AKT pathway is activated, through the presence of the HGF (Hepatocyte Growth Factors), EGF (Epidermal Growth Factors) and FGF (Fibroblast Growth Factors) ligands, it leads to a proliferative phenotype. The activation of this pathway results in the activation of PDPK1 and mTOR, both able to phosphorylate p70 (RPS6KB1) which then promotes cell proliferation and growth (Consortium 2019). There has been some evidence of negative regulations of these two pathways carried out by ERK itself (Lake, Corrêa, and Müller 2016): phosphorylated ERK is able to prevent the SOS-GRB2 complex formation through the activation of SPRY (Edwin et al. 2009), inhibit the EGF-dependent GAB1/PI3K association (Lehr et al. 2004) and down-regulate EGFR signal through phosphorylation (Lake, Corrêa, and Müller 2016). The model also accounts for a negative regulation of proliferation through a pathway involving p53 activation in response to DNA damage (represented by ATM); p53 hinders proliferation through the activation of both PTEN, a PI3K inhibitor, and p21 (CDKN1A) responsible for cell cycle arrest.
We hypothesize that a single network is able to discriminate between melanoma and CRC cells. These differences may come from different sources. One of them is linked to the negative feedback loop from ERK to EGFR. As mentioned previously, this feedback leads to one important difference in response to treatment between melanoma and CRC: \(BRAF^{(V600E)}\) inhibition causes a rapid feedback activation of EGFR, which supports continued proliferation. This feedback is observed only in colorectal since melanoma cells express low levels of EGFR and are therefore not subject to this reactivation (Prahallad et al. 2012). Moreover, phosphorylation of SOX10 by ERK inhibits its transcription activity towards multiple target genes by interfering with the sumoylation of SOX10 at K55, which is essential for its transcriptional activity (Han et al. 2018). The absence of ERK releases the activity of SOX10, which is necessary and sufficient for FOXD3 induction. FOXD3 is then able to directly activate the expression of ERBB3 at the transcriptional level, enhancing the responsiveness of melanoma cells to NRG1 (the ligand for ERBB3), and thus leading to the reactivation of both MAPK and PI3K/AKT pathways (Han et al. 2018). Furthermore, it has been shown that in colorectal cells, FOXD3 inhibits EGFR signal in vitro (Li et al. 2017). Interestingly, SOX10 is highly expressed in melanoma cell lines when compared to other cancer cells. In the model, we define SOX10 as an input because of the lack of information about the regulatory mechanisms controlling its activity. The different expression levels of SOX10 have been reported to play an important role in melanoma (high expression) and colorectal (low expression) cell lines.
Besides a list of formalized biological assertions, retrieved from literature, has been used during the model building to ensure the consitency of the model with some qualitative behaviours. These assertions, listed below, are all verified when the logical model is simulated (details are available on the corresponding GitHub repository):
- BRAF inhibition causes a feedback activation of EGFR in colorectal cancer and not in melanoma (Prahallad et al. 2012)
- MEK inhibition stops ERK signal but activates the PI3K/Akt pathway and increases the activity of ERBB3 (Gopal et al. 2010; Lake, Corrêa, and Müller 2016)
- HGF signal leads to the reactivation of the MAPK and PI3K/AKT pathways, and resistance to BRAF inhibition (Wroblewski et al. 2013)
- BRAF inhibition in melanoma activates the SOX10/FOXD3/ERBB3 axis, which mediates resistance through the activation of the PI3K/AKT pathway (Han et al. 2018)
- Overexpression/mutation of CRAF results in constitutive activation of ERK and MEK also in the presence of a BRAF inhibitor [Manzano et al. (2016); johannessen2010cot]
- Early resistance to BRAF inhibition may be observed in case of PTEN loss, or mutations in PI3K or AKT (Manzano et al. 2016)
- Experiments in melanoma cell lines support combined treatment with BRAF/MEK + PI3K/AKT inhibitors to overcome resistance (Manzano et al. 2016)
- BRAF inhibition (Vemurafenib) leads to the induction of PI3K/AKT pathway and inhibition of EGFR did not block this induction (Corcoran et al. 2012)
- Induction of PI3K/AKT pathway signaling has been associated with decreased sensitivity to MAPK inhibition (Corcoran et al. 2012)
B.4 Logical model of prostate cancer
In the context of the European project PRECISE (Personalized Engine for Cancer Integrative Study and Evaluation), focused on the integrative study of prostate cancer, an adapted logical model has been built. This prostate cancer model is initially based on the generic structure of the Fumia model presented in section B.1, which has been considerably enriched and extended with genes and mechanisms specific to prostate cancer such as ERG, SPOP or AR. The model contains 133 nodes and 449 edges (Figure B.3) and includes pathways like androgen receptor and growth factor signalling, several signaling pathways (Wnt, NFkB, PI3K/AKT, MAPK, mTOR, SHH), cell cycle, epithelial-mesenchymal transition (EMT), Apoptosis, DNA damage, etc. The model has 9 inputs (EGF, FGF, TGF beta, Nutrients, Hypoxia, Acidosis, Androgen, TNF alpha and Carcinogen presence) and 6 outputs (Proliferation, Apoptosis, Invasion, Migration, (bone) Metastasis and DNA repair).
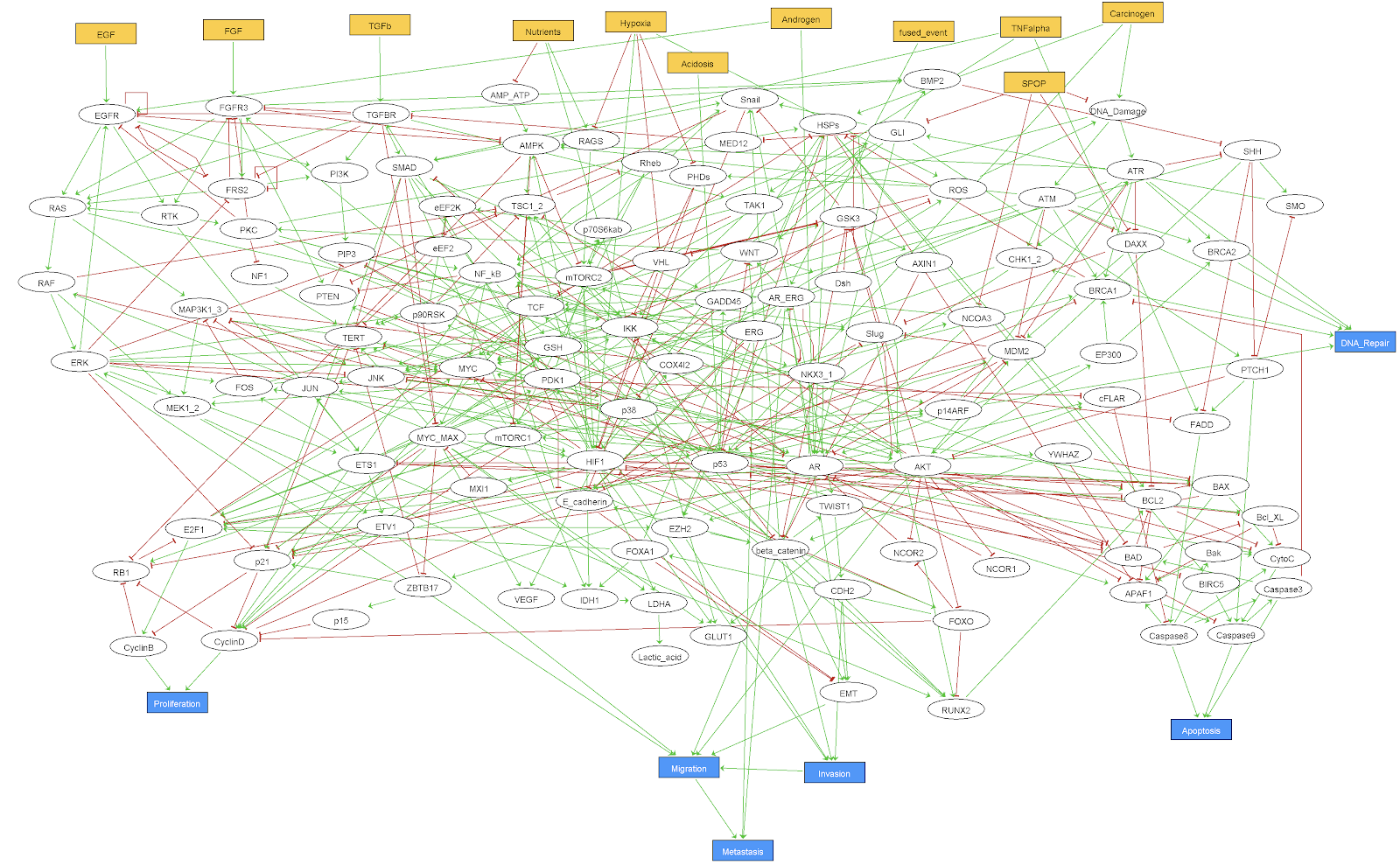
Figure B.3: GINsim representation of the 'Montagud' logical model of prostate cancer.
References
Consortium, UniProt. 2019. “UniProt: A Worldwide Hub of Protein Knowledge.” Nucleic Acids Research 47 (D1). Oxford University Press: D506–D515.
Corcoran, Ryan B, Hiromichi Ebi, Alexa B Turke, Erin M Coffee, Michiya Nishino, Alexandria P Cogdill, Ronald D Brown, et al. 2012. “EGFR-Mediated Reactivation of Mapk Signaling Contributes to Insensitivity of Braf-Mutant Colorectal Cancers to Raf Inhibition with Vemurafenib.” Cancer Discovery 2 (3). AACR: 227–35.
Edwin, Francis, Kimberly Anderson, Chunyi Ying, and Tarun B Patel. 2009. “Intermolecular Interactions of Sprouty Proteins and Their Implications in Development and Disease.” Molecular Pharmacology 76 (4). ASPET: 679–91.
Fumia, Herman F, and Marcelo L Martins. 2013. “Boolean Network Model for Cancer Pathways: Predicting Carcinogenesis and Targeted Therapy Outcomes.” PloS One 8 (7). Public Library of Science.
Gopal, YN Vashisht, Wanleng Deng, Scott E Woodman, Kakajan Komurov, Prahlad Ram, Paul D Smith, and Michael A Davies. 2010. “Basal and Treatment-Induced Activation of Akt Mediates Resistance to Cell Death by Azd6244 (Arry-142886) in Braf-Mutant Human Cutaneous Melanoma Cells.” Cancer Research 70 (21). AACR: 8736–47.
Han, Shujun, Yibo Ren, Wangxiao He, Huadong Liu, Zhe Zhi, Xinliang Zhu, Tielin Yang, et al. 2018. “ERK-Mediated Phosphorylation Regulates Sox10 Sumoylation and Targets Expression in Mutant Braf Melanoma.” Nature Communications 9 (1). Nature Publishing Group: 1–14.
Lake, David, Sonia AL Corrêa, and Jürgen Müller. 2016. “Negative Feedback Regulation of the Erk1/2 Mapk Pathway.” Cellular and Molecular Life Sciences 73 (23). Springer: 4397–4413.
Lehr, Stefan, Jorg Kotzka, Haluk Avci, Albert Sickmann, Helmut E Meyer, Armin Herkner, and Dirk Muller-Wieland. 2004. “Identification of Major Erk-Related Phosphorylation Sites in Gab1.” Biochemistry 43 (38). ACS Publications: 12133–40.
Li, Kun, Qunfeng Guo, Jun Yang, Hui Chen, Kewen Hu, Juan Zhao, Shunxin Zheng, et al. 2017. “FOXD3 Is a Tumor Suppressor of Colon Cancer by Inhibiting Egfr-Ras-Raf-Mek-Erk Signal Pathway.” Oncotarget 8 (3). Impact Journals, LLC: 5048.
Manzano, José Luís, Laura Layos, Cristina Bugés, María de los Llanos Gil, Laia Vila, Eva Martinez-Balibrea, and Anna Martínez-Cardús. 2016. “Resistant Mechanisms to Braf Inhibitors in Melanoma.” Annals of Translational Medicine 4 (12). AME Publications.
Prahallad, Anirudh, Chong Sun, Sidong Huang, Federica Di Nicolantonio, Ramon Salazar, Davide Zecchin, Roderick L Beijersbergen, Alberto Bardelli, and René Bernards. 2012. “Unresponsiveness of Colon Cancer to Braf (V600e) Inhibition Through Feedback Activation of Egfr.” Nature 483 (7387). Nature Publishing Group: 100–103.
Verlingue, Loic, Laurence Calzone, Maud Kamal, Nicolas Servant, Lisa Belin, Emmanuel Barillot, and Christophe Le Tourneau. 2016. “In Silico Prediction of the Clinical Response to the MTOR Inhibitor Everolimus Using a Boolean Model: Validation from a Cohort of the Shiva Trial.” AACR.
Verlingue, Loic, Aurélien Dugourd, Gautier Stoll, Emmanuel Barillot, Laurence Calzone, and Arturo Londoño-Vallejo. 2016. “A Comprehensive Approach to the Molecular Determinants of Lifespan Using a Boolean Model of Geroconversion.” Aging Cell 15 (6). Wiley Online Library: 1018–26.
Wellbrock, Claudia, Maria Karasarides, and Richard Marais. 2004. “The Raf Proteins Take Centre Stage.” Nature Reviews Molecular Cell Biology 5 (11). Nature Publishing Group: 875–85.
Wroblewski, David, Branka Mijatov, Nethia Mohana-Kumaran, Fritz Lai, Stuart J Gallagher, Nikolas K Haass, Xu Dong Zhang, and Peter Hersey. 2013. “The Bh3-Mimetic Abt-737 Sensitizes Human Melanoma Cells to Apoptosis Induced by Selective Braf Inhibitors but Does Not Reverse Acquired Resistance.” Carcinogenesis 34 (2). Oxford University Press UK: 237–47.